News
Breaking Discovery: NAV3 Gene Linked to a Novel Neurodevelopmental Disorder (NEDUA) – OMIM #621182
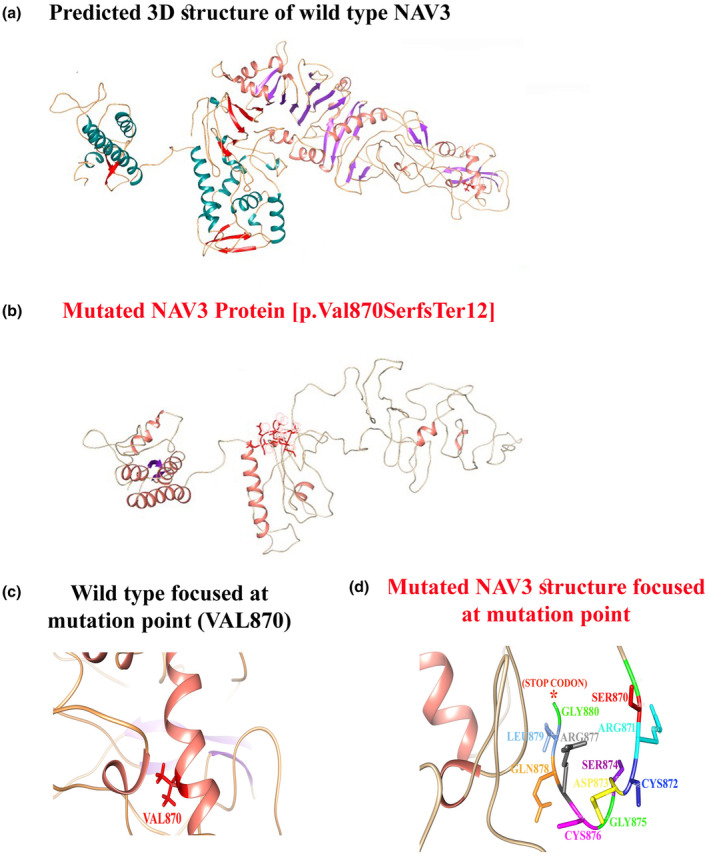
We are pleased to share a significant milestone in the field of neurogenetics. The Editor in Chief of the Journal of Biochemical and Clinical Genetics, Prof. Majid Alfadhel, along with his team, has recently identified a novel gene-disease association involving NAV3, which has now been officially recognized by OMIM as “Umair-Alfadhel Neurodevelopmental Disorder (NEDUA)” [OMIM #621182] — named in honor of Dr. Muhammad Umair (Editorial Board Member) and Prof. Majid Alfadhel (Editor in Chief) for their pioneering contribution.
Key Findings:
- Gene: NAV3 (Neuron Navigator 3), crucial for neuronal morphogenesis and axonal guidance.
- Clinical Features: Patients present with global developmental delay, poor or absent speech, dysmorphic facies, microcephaly, hypotonia, and additional neurodevelopmental features.
- Inheritance: Autosomal recessive, with both homozygous and compound heterozygous variants identified in affected families.
- Impact: This discovery provides much-needed answers for families worldwide and opens new avenues for research into targeted therapies and genetic counseling.
Read More:
• OMIM #621182
• Umair et al., 2024 [PubMed: 39038237]
This achievement notably marks the second time where a disease has been named after Prof. Alfadhel, following the recognition of Alfadhel Syndrome, which is registered in the Online Mendelian Inheritance in Man (OMIM) database with the identifier #620655. Prof. Alfadhel is the second scientist from Saudi Arabia to have two diseases named after him in the OMIM database.
Let’s continue to advance our understanding of neurodevelopmental disorders together!
Announcements

Technical Alert
Dear authors, reviewers, and visitors, The Journal of Biochemical and Clinical Genetics (JBCGenetics) is currently in the process of migrating to a new server and a new online journal submission and publishing system (epublisyst.com). You may experience temporary site outages, broken links, or other technical issues. We kindly ask for your patience during this transition, and we will be back online as soon as possible.
If you had submitted a manuscript through our previous submission system (eJmanager), you can continue to check for updates by logging into the previous journal submission system:
https://www.ejmanager.com/my/jbcg/
Reviewers who accepted to review via our previous journal system (eJmanager), may log in via following link:
https://www.ejmanager.com/reviewers/index.php?isl=login
Thank you for your understanding and continued support.


